

Le blog participatif de bioinformatique francophone depuis 2012
Qui sommes nous ?

Une communauté d'étudiants et de professionnels

Une base d'articles libres pour tous niveaux

Des salons de discussion actifs au quotidien
-
Synchronisez vos PDF Zotero sur Android
Après un an d'errance, j'ai enfin réussi à synchroniser ma collection d'articles en PDF entre mon pc et ma liseuse, et surtout les annotations faites depuis ma liseuse. Bon, j'avais lâché l'affaire pendant longtemps, mais un message de @sebgra sur notre serveur Discord m'a fait remettre les mains dans le cambouis. Comme vous le savez,…
-
IA et bioinformatique : exploitons les réseaux convolutionnels (CNN)

Interessons-nous aujourd'hui aux séquences d’ADN. Nous utiliserons le dataset téléchargeable ici : https://www.kaggle.com/datasets/nageshsingh/dna-sequence-dataset L'ensemble des fichiers nécessaire à cet article sont disponibles ici. Vous trouverez dans ce lot de données un ensemble de séquences d’ADN issues de 3 espèces : l’homme, le chien et le chimpanzé. Chacune de ces séquences appartient à une des 7 familles de…
-
Licence double "Informatique — Sciences de la vie" à Évry

La licence double Informatique — Sciences de la vie est une formation double diplôme de l'Université Paris — Saclay dispensée à la fois à l'Université de Versailles Saint-Quentin et à l'Université d'Évry val d'Essonne. Cette formation en trois ans vise à apporter aux étudiants des compétences solides dans les deux domaines que sont la biologie…
-
Qu'est-ce que la toxicogénomique ?
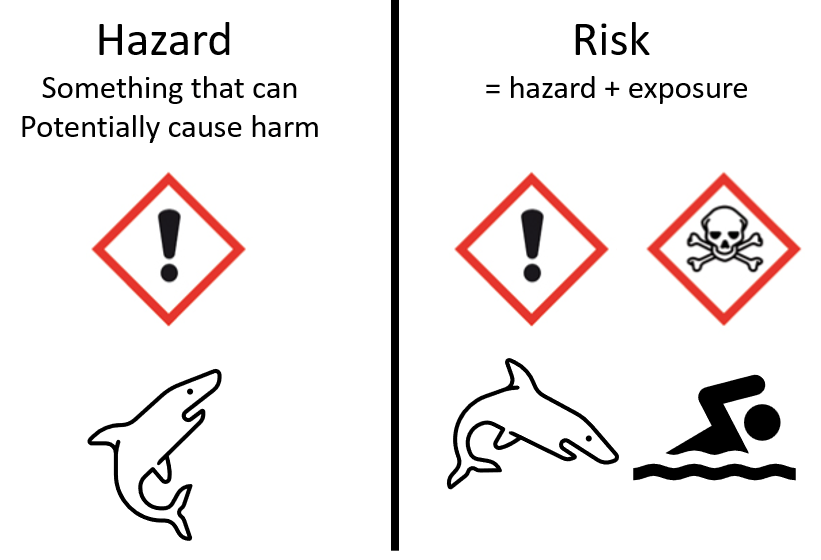
Oyez, oyez humble lecteur, aujourd'hui je vous propose de partir en voyage dans le monde de la toxicologie. À quoi peut bien servir les données OMICS pour un toxicologue ? Hé bien, à faire de la toxicogénomique pardi ! Dans ce billet je vous propose une petite porte sur ce domaine d'application des omics et de se…
-
Bonnes fêtes de fin d'année !

Chers bioinfo-freurs et bioinfo-freuses, nous vous souhaitons de très bonne fêtes ! Que cette fin d'année soit aussi douce et agréable qu'une bonne tasse de chocolat chaud au coin du feu. De nôtre côté, nous disons au revoir avec la larmichette à l'oeil à notre co-fondateur et administrateur historique Yoann Mouscaz, qui part voguer sous d'autres…
-
Pourquoi je ne lis pas tous les CVs de la même manière

Dans un précédent billet je me suis laissé vagabonder autour de l'idée qu'un bioinformaticien était un data truc comme un autre. Dans ces lignes je vais exprimer pourquoi il est important à mon sens de bien travailler son CV… et ce qui personnellement me fait réfléchir sur la forme de CV liée à nos métiers.…
-
Les mystérieuses cités d'or

La bioinformatique est un domaine de recherche donc, avec régulièrement de nouveaux logiciels et très souvent* votre premier contact avec ce nouveau logiciel se fait via la forge logiciel GitHub. Sur GitHub la première chose que l'utilisateur voit c'est le Readme (pour plus de détails sur ce fichier aller lire cet excellent billet de blog).…
-
Bioinformatique et IA : un premier pas

Intelligence Artificielle, Machine Learning, Deep-Learning, quid du Data-Scientist Intelligence artificielle (IA), Machine learning (Apprentissage machine, pour les francophones), Deep-learning (Apprentissage profond), autant de termes si étrangers et familiers à la fois… Comment se retrouver dans cette jungle de termes techniques ? Commençons par définir ce qu'est l'IA. Base de science-fiction pour certains, source d'inquiétudes pour d'autres,…
-
De bioinformaticien à data scientist, un simple pas ?
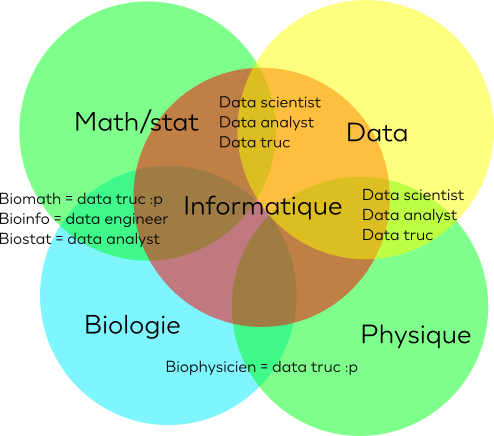
Nombreux parmi nous se retrouvent un jour en fin de master/thèse ou postdoc de bioinformatique. A ce moment là, dans les difficultés de la recherche d'emplois on vient à se demander : "Et si je postulais à toutes ces offres de data scientist ?"Dans ces lignes, je vais donner mon avis de manière libre, sur pourquoi…